新药研发:获批临床的抗肿瘤1类新药醋酸吡可利
前言
近日,由浙江大学和上海药物所联合自主研发的抗肿瘤1类新药醋酸吡可利布(PY34AC)获批临床试验。该候选分子为细胞周期检查点激酶1(CHK1)高选择性的抑制剂,目前与海门百极弘烨医药科技有限公司合作开展临床试验。

PY34AC分子结构暂不能完全确定为上述分子,目前该项目仅于2019年在European Journal ofMedicinal Chemistry发表了最优化合物(R)-17的发现,本文将基于该分子了解该项目的基础研究过程。
CHK1抑制剂的研发已久,第一代的抑制剂存在CHK1/2选择性差、脱靶导致副作用明显的问题,目前除了礼来的LY,均已终止临床试验。第二代具有高选择性的CHK1抑制剂已有多个分子进入临床研究阶段。
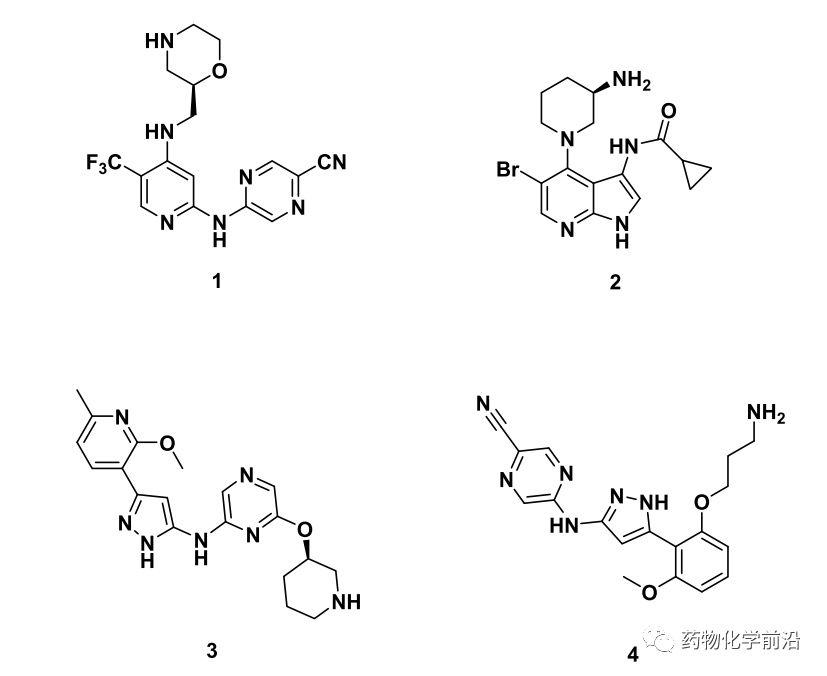
为发现具有新骨架的CHK1抑制剂,本项目通过虚拟筛选的方法从Chembridge数据库及In-houselibrary共分子中筛选出10个hit。其中MCL1020是唯一来自In-houselibrary的化合物,同时该分子可看作已有抑制剂SRA-737(上图化合物1)骨架跃迁而来。

MCL1020对接到CHK1如下图,二氨基嘧啶骨架与常见的激酶抑制剂结合方式一致,氰基与Lys38可以形成氢键相互作用,基于该对接结果进行了一系列的结构优化。
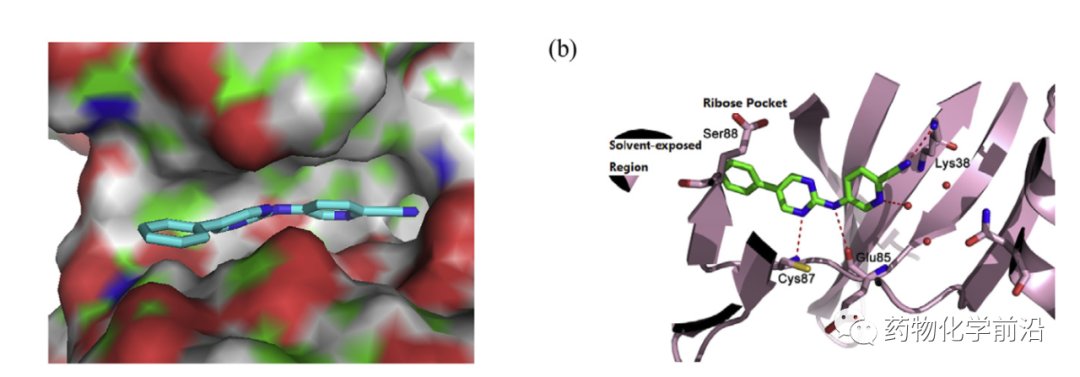
对接发现MCL1020的C环暴露于溶剂区域边缘,B环也可引入较大的取代基团,因此首先在C环和B环空腔位置引入极性基团,如下表,其中在B环引入后(化合物6)活性显著提升,而C环引入活性不佳。故该将化合物6为新的lead进一步优化,C环不同取代发现甲基吡咯取代的化合物15具有最优活性。
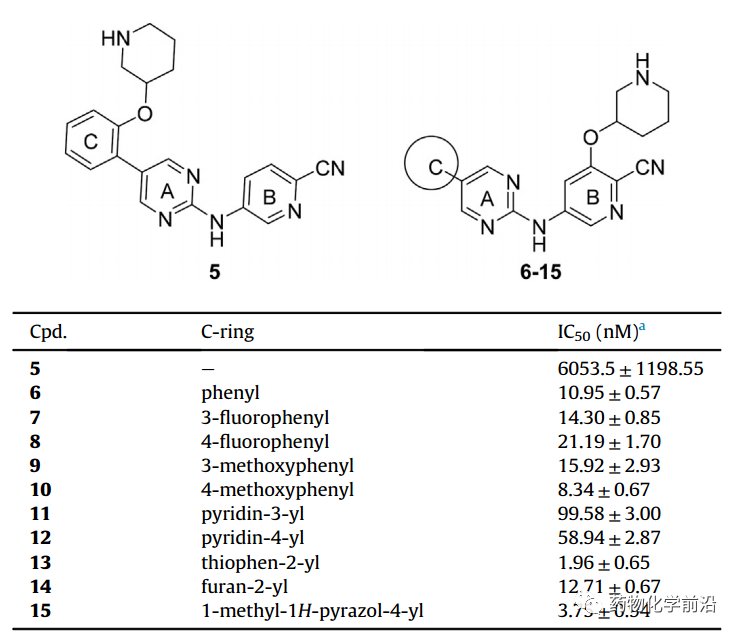
以化合物15为基础进一步优化,对接发现MCL1020的A环仍有一定空腔,可设计小基团的取代,因而在A环4位引入甲氧基或甲氨取代,发现甲氨取代(化合物17)具有显著活性,而甲氧基取代则活性明显丧失。固定甲氨取代进一步优化C环,发现化合物17(甲基吡咯取代)具有最优活性。
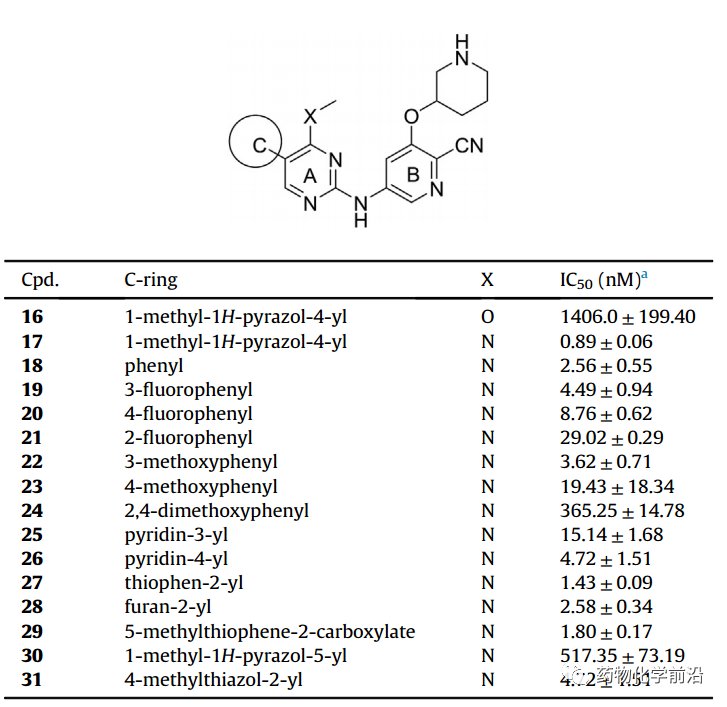
最后对B环的极性基团进行优化,最终发现R构型取代的化合物具有更优活性,NH对活性影响明显,确定最优化合物(R)-17与当前临床阶段分子具有相当的酶活活性,进行进一步的研究。
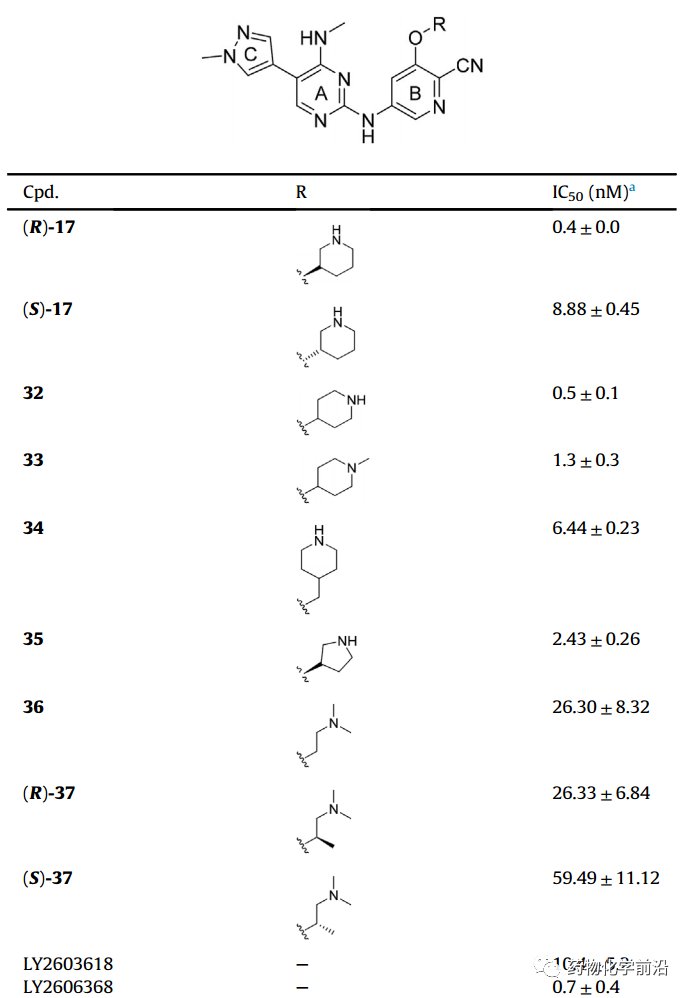
细胞水平化合物(R)-17在Z-138细胞株表现出最优活性,与临床分子LY相当。
同时化合物(R)-17在CHK2上具有非常高的选择性,在CDKs、PIMs等靶点上也有明显的选择性,显著优于LY。
通过移植瘤模型评价,(R)-17在10mg/kg的注射剂量下抑瘤率能达到78.64%,20mg/kg抑瘤率为90.29%。
通过hERG抑制毒性评价发现化合物(R)-17显著优于临床分子LY和LY。
总结:
从本文报道前期研究过程可以清晰的看到该项目从Hit—lead—candidate的全过程。本文发现的分子酶活、细胞活性和当前的CHK1抑制剂相当,但其显著提高了选择性,并降低了毒性。
参考文献:
[1] Tianhua Zhang. Discovery of(R)-5-((5-(1-methyl-1H-pyrazol-4-yl)-4-(methylamino)pyrimidin-2-yl)amino)-3-(piperidin-3-yloxy)picolinonitrile,a novel CHK1 inhibitor for hematologic malignancies. Eur. J. 173 (2019) 44-62.
【来源:健康界】
声明:转载此文是出于传递更多信息之目的。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。